

Our Team Explains: Mosaicism
Mosaicism is a biological phenomenon in which a person has two or more genetically different sets of cells. Although mosaicism may have no effect, it could also lead to genetic disorders and even cau..



Mosaicism is a biological phenomenon in which a person has two or more genetically different sets of cells. Although mosaicism may have no effect, it could also lead to genetic disorders and even cau..
Background information on in vitro diagnostic services Laboratory-based testing methods and medical devices play a critical role in diagnosis and medical decision-making. Indeed, different tests, ..

Traditional DNA tests may overlook 10% of classic in Familial Adenomatous Polyposis (FAP) cases. By integrating RNA sequencing, researchers unveiled novel APC variants in six families, explaining mis..
Overview In January 2024, the American Society of Clinical Oncology (ASCO) and the Society of Surgical Oncology (SSO) published new recommendations for hereditary breast cancer genetic testing (1)..

Endometriosis is a chronic gynecological condition that affects 1 in 10 women of reproductive age worldwide [1]. It can manifest with the first menstruation and last until menopause. As March has bee..

Rare Disease Day is a global awareness day held annually to raise awareness of all rare diseases. It was first celebrated in 2008, on the rarest day – February 29. Since then, it is celebrated each..
Cancer is a group of genetic diseases that can develop almost anywhere in the body. Many people in the world are affected by cancer every year. Following our video that explains the basics of cancer,..

Movember is all about men’s health
The month of November, also known as Movember, has been chosen as the month to raise awareness about men’s health. During Movember, men are encouraged to grow a moustache, or participate in an even..
Health and well-being
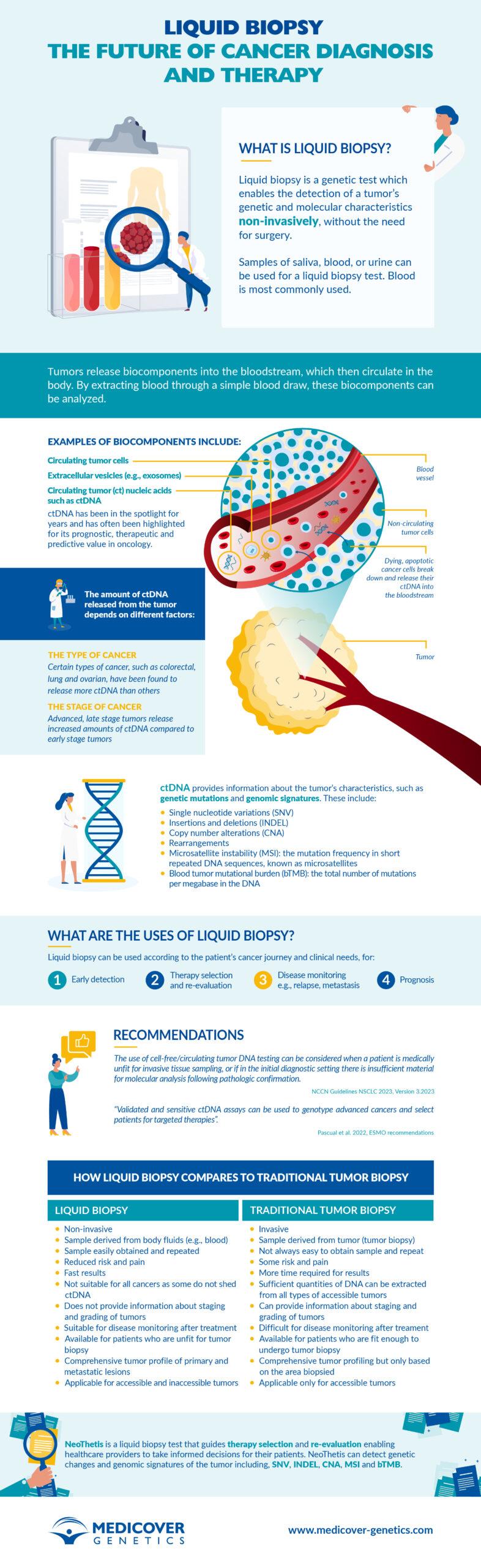
Liquid biopsy: a game-changer for cancer diagnosis and therapy
Liquid biopsy is a novel, non-invasive blood test that enables healthcare providers to discover a range of information about a tumor, such as early cancer detection, genetic alterations that lead to ..
Hereditary diseases

Who Is The Father of Genetics?
The father of genetics is Gregor Mendel. Mendel was an Austrian monk, whose experiments breeding pea plants in the monastery garden led to breakthroughs in our understanding of genetics and heredity…
All about genetics
Movember is all about men’s health
The month of November, also known as Movember, has been chosen as the month to raise awareness about men’s health. During Movember, men are encouraged to grow a moustache, or participate in an even..
Health and well-being
Liquid biopsy: a game-changer for cancer diagnosis and therapy
Liquid biopsy is a novel, non-invasive blood test that enables healthcare providers to discover a range of information about a tumor, such as early cancer detection, genetic alterations that lead to ..
Hereditary diseases
Who Is The Father of Genetics?
The father of genetics is Gregor Mendel. Mendel was an Austrian monk, whose experiments breeding pea plants in the monastery garden led to breakthroughs in our understanding of genetics and heredity…
All about genetics
Our editors are a diverse group of scientists and physicians specialized in different areas of genetics and genetic testing technologies. Owing to our various backgrounds, our editors represent a broad range of expertise and are responsible for the content shared on our website. Only evidence-based, peer-reviewed, and up-to-date content is shared, and all text is continuously edited to maximize its correctness.


Medicover Genetics’ technology transfer platform offers comprehensive testing in a single sequencing run, making it a unique and efficient solution for laboratories of any size. With our Technology Transfer platform, laboratories can perform multi-disciplinary genetic tests such as NIPT, cardiac, hereditary cancer, carrier screening, and metabolic testing all in a single sequencing run. This improves turnaround time and reduces operational costs while ensuring high-quality results with no compromise on accuracy. Our comprehensive testing approach is ideal for laboratories looking to provide a wide range of genetic tests to their healthcare associates and patients.
Our Technology Transfer platform workflow is designed to minimize the risk of errors, reduce hands-on processing and increase the accuracy of test results, providing confidence to patients and physicians in making informed decisions with our user-friendly protocols. The implementation of these protocols ensures that laboratory personnel are trained to follow standardized procedures, reducing the likelihood of human error, and increasing the quality of results.
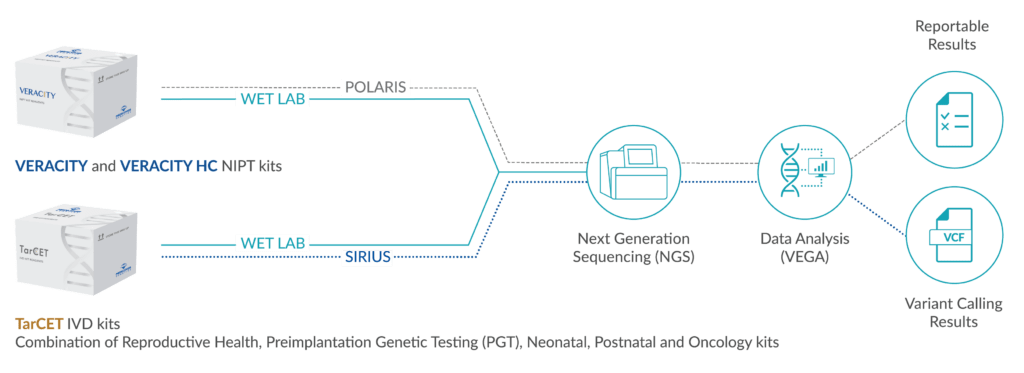
Medicover Genetics’ versatile Technology Transfer platform can be integrated in a single workflow, offering a multidisciplinary portfolio of genetic tests. VERACITY, VERACITY High Content (HC) and TarCET IVD kits are CE-marked and include user friendly color identification formatting. VERACITY and VERACITY HC kits contain 96 reactions and TarCET IVD kits contain 16 reactions.



We work closely with laboratories to meet their unique needs and goals. We understand that every laboratory is different, and we tailor our approach to accommodate the specific requirements of each partner. In collaboration with our partners, we design a seamless integrated process, from initial setup to ongoing support and maintenance. Our experienced team provides technical, commercial, and marketing assistance to ensure a successful partnership and optimal performance. We aim to build long-lasting partnerships that are mutually beneficial.
